Dynamic transformers#
In conventional chemometrics practice, preprocessing is treated as a static operation: a correction is determined from a calibration set and then applied uniformly to every new spectrum. The preprocessing step holds everything it needs — a stored mean spectrum, a fitted baseline, a set of PLS loadings — and the data alone is sufficient at prediction time.
This works well for a large class of problems, but it breaks down when the correction you need to apply depends on information that is only available at inference time. In practice this information falls into two categories:
Measurement metadata — instrument-level quantities recorded alongside the spectrum but not part of it: the x-axis calibration, laser power, integration time, detector temperature.
Process data — sample- or batch-level context that changes between runs or samples: a fresh background measurement, a dilution factor, a reference standard collected just before the sample or other process parameters such as temperature or humidity.
Some concrete examples from spectroscopy:
The x-axis grid of the instrument drifted between calibration and deployment. Each new spectrum arrives on a slightly different wavenumber array.
You want to normalize by laser power or integration time, values that are logged per measurement but are not part of the spectrum itself.
A background spectrum is measured fresh before each sample batch and must be subtracted at inference, not at fit time.
chemotools addresses this with a set of dynamic transformers — estimators
that accept additional per-call parameters alongside X at transform
time, delivered through scikit-learn's metadata routing framework.
Transformer |
Metadata argument |
Use case |
|---|---|---|
|
|
Align spectra measured on different wavenumber grids |
|
|
Normalize by laser power, integration time, temperature factor |
|
|
Subtract a freshly measured background at inference |
Example: aligning spectra to a common grid#
In Raman spectroscopy, each instrument has a slightly different pixel-to-wavenumber calibration. Spectra from different instruments share the same chemistry but arrive on different x-axis grids — so they cannot be stacked into a matrix until they are resampled onto a common one.
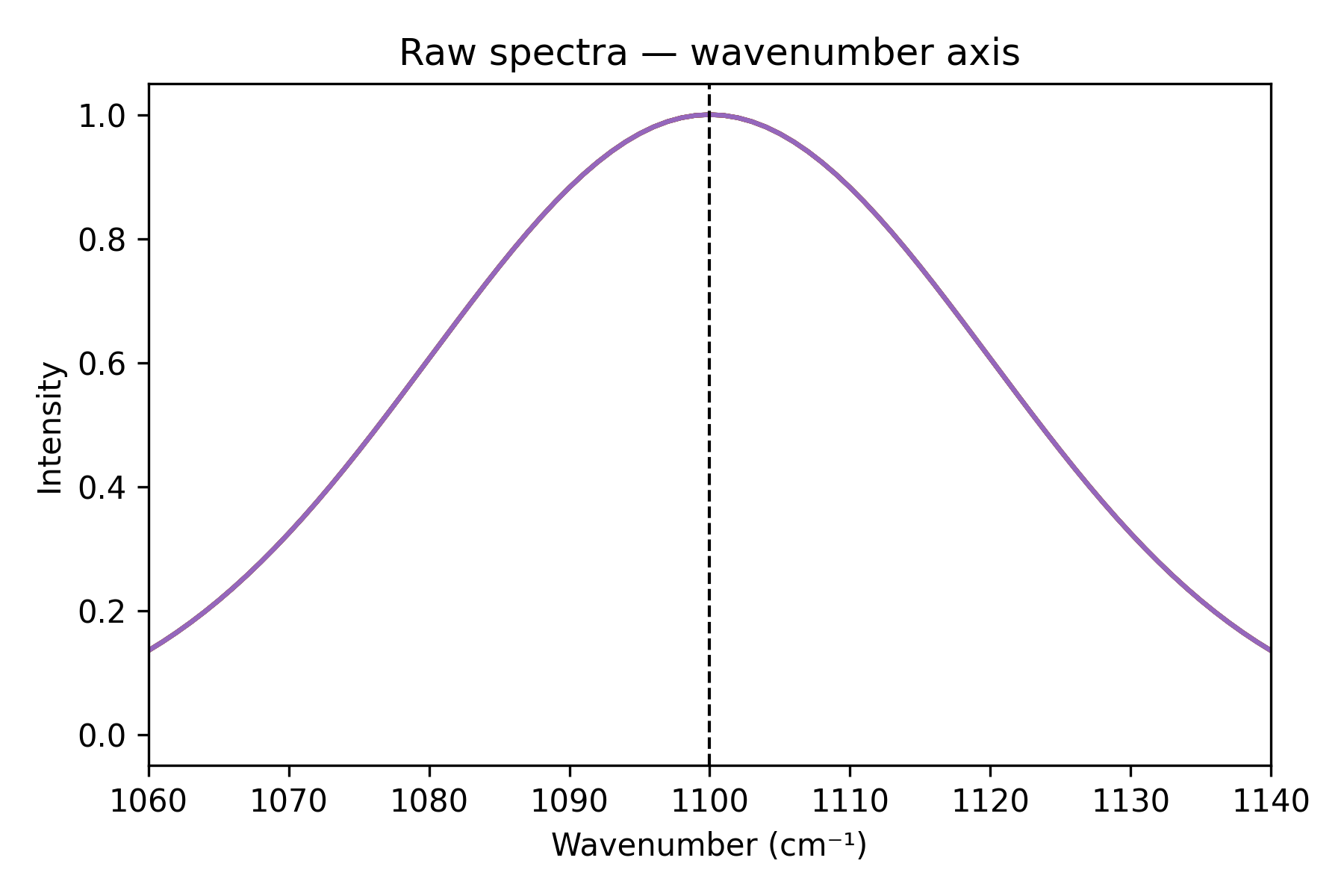
Five simulated spectra, each with a Gaussian peak at 1100 cm⁻¹ but on a slightly different grid, illustrate the problem.
Setting up the data
import numpy as np
import sklearn
import matplotlib.pyplot as plt
from chemotools.adaptation import XAxisInterpolator
sklearn.set_config(enable_metadata_routing=True) # explained in "How metadata routing works" below
N = 1000 # pixels per spectrum
sigma = 20 # peak width (pixels)
offsets = [-10, -5, 0, 5, 10] # pixel-grid offset per instrument
raw_spectra, raw_x_axes = [], []
for offset in offsets:
peak = N // 2 + offset
y = np.exp(-0.5 * ((np.arange(N) - peak) / sigma) ** 2)
x = np.arange(N) + (1100 - peak) # x[peak] == 1100 wn
raw_spectra.append(y)
raw_x_axes.append(x)
raw_spectra = np.array(raw_spectra) # shape (5, 1000)
raw_x_axes = np.array(raw_x_axes) # shape (5, 1000)
Step 1 — what the instrument gives you
Each spectrum is delivered as an array of intensity values indexed by pixel number. When you plot them on a common pixel axis, the peaks appear at different positions — each instrument's zero point is slightly different.
zoom = 40
fig, ax = plt.subplots(figsize=(6, 4))
for y in raw_spectra:
ax.plot(y)
ax.set_xlim(N // 2 - zoom, N // 2 + zoom)
ax.set(title="Raw spectra — pixel index", xlabel="Pixel index", ylabel="Intensity")
plt.tight_layout()
plt.show()
Peaks land at different pixel positions — the grids are misaligned. If you stacked these rows into a matrix as-is and fed it to a PLS model, column k would represent a different wavenumber for each instrument, so every learned regression coefficient would point at the wrong feature.
Step 2 — plot against wavenumber
Each spectrum comes with its own wavenumber axis. Plotting against it shows the peaks coincide at 1100 cm⁻¹, but the arrays are still all different.
fig, ax = plt.subplots(figsize=(6, 4))
for y, x in zip(raw_spectra, raw_x_axes):
ax.plot(x, y)
ax.axvline(1100, color="k", linestyle="--", linewidth=1)
ax.set_xlim(1100 - zoom, 1100 + zoom)
ax.set(title="Raw spectra — wavenumber axis", xlabel="Wavenumber (cm⁻¹)", ylabel="Intensity")
plt.tight_layout()
plt.show()
Step 3 — interpolate onto a common grid
XAxisInterpolator takes a common_x_axis
defined once at construction time and, at every transform call, resamples
each row from its own x_axis onto that shared grid. The per-spectrum
axis is passed as metadata — not baked into the transformer — so it can
change freely between calls.
x_common = np.linspace(650, 1550, N)
interpolator = (
XAxisInterpolator(
common_x_axis=x_common, method="linear", left=0, right=0
) # left/right fill values outside the grid
.set_fit_request(x_axis=True)
.set_transform_request(x_axis=True)
)
aligned_spectra = interpolator.fit_transform(raw_spectra, x_axis=raw_x_axes)
fig, ax = plt.subplots(figsize=(6, 4))
for y in aligned_spectra:
ax.plot(y)
ax.set(
title="Aligned spectra — common-axis index",
xlabel="Common-axis index",
ylabel="Intensity",
)
plt.tight_layout()
ax.set_xlim(420, 580)
plt.show()
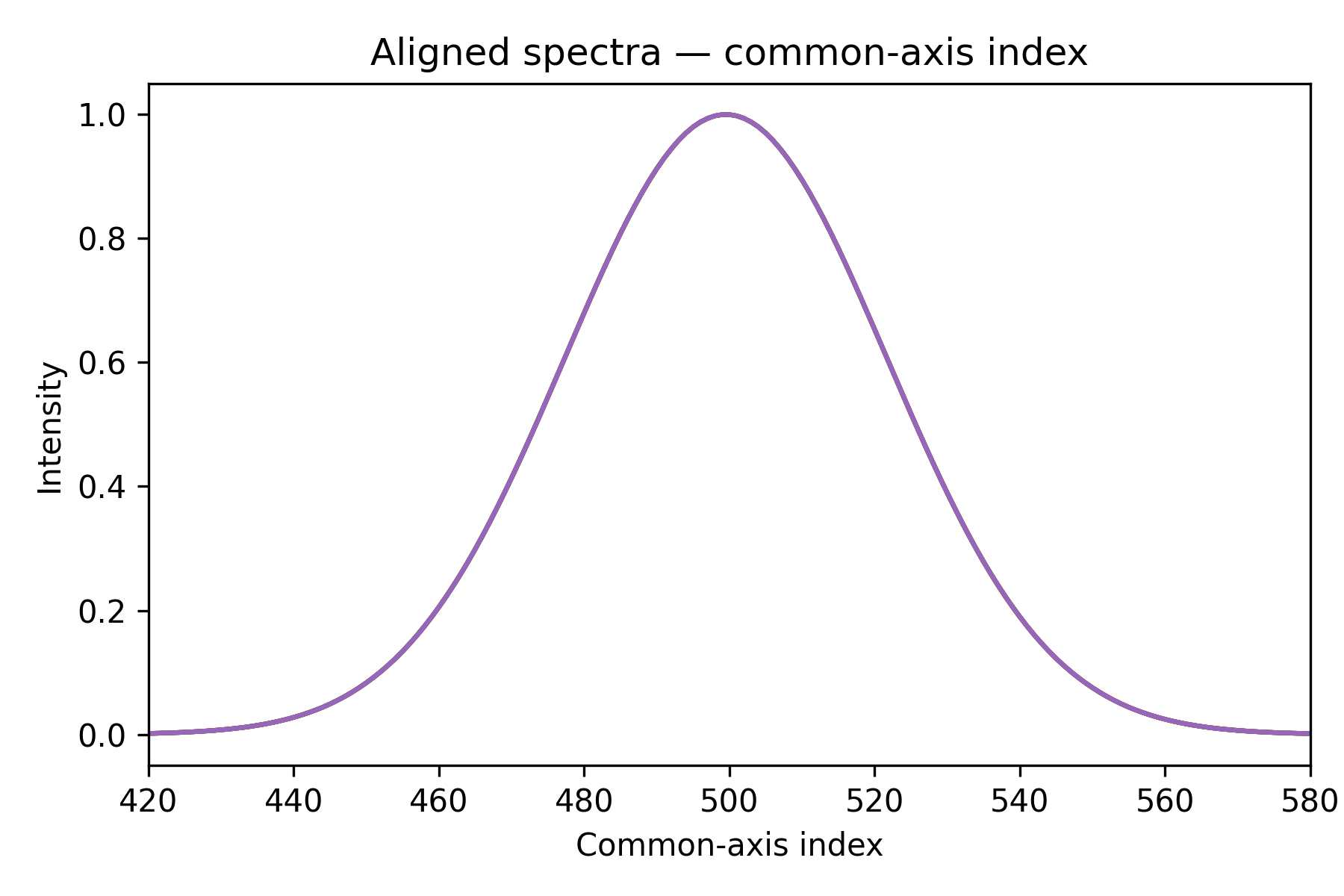
All five peaks now sit at the same column index. The matrix aligned_spectra
can be fed directly into any subsequent step or model.
How metadata routing works#
The two method calls on the interpolator — set_fit_request(x_axis=True)
and set_transform_request(x_axis=True) — register x_axis as a
metadata argument for the fit and transform phases respectively.
When you pass x_axis to a pipeline call, scikit-learn delivers it only
to the step that declared it; every other step is unaffected.
set_fit_request covers fit and fit_transform;
set_transform_request covers transform. Both are declared in the
example because a Pipeline calling
fit_transform routes metadata through both phases.
Using it inside a Pipeline#
The pipeline below continues from the same variables defined above:
from sklearn.pipeline import Pipeline
from sklearn.preprocessing import StandardScaler
from chemotools.scatter import MultiplicativeScatterCorrection
pipe = Pipeline(
[
(
"interpolate",
XAxisInterpolator(
common_x_axis=x_common, method="linear", left=0, right=0
) # left/right fill values outside the grid
.set_fit_request(x_axis=True)
.set_transform_request(x_axis=True),
),
("msc", MultiplicativeScatterCorrection()),
("scaler", StandardScaler()),
]
)
# x_axis is routed to "interpolate" only; the other steps never see it
X_preprocessed = pipe.fit_transform(raw_spectra, x_axis=raw_x_axes)
注釈
Only the step that declared set_transform_request(x_axis=True) receives
x_axis. The other steps in the pipeline are unaffected.
Interpolation methods#
XAxisInterpolator supports three methods,
selectable via the method parameter:
|
Description |
When to use |
|---|---|---|
|
Piecewise linear interpolation. |
Fast; best when spectra are smooth and grids are closely spaced. |
|
Natural cubic spline (via |
Good all-round choice; smooth and accurate. |
|
Piecewise cubic Hermite (via |
Preserves monotonicity; avoids overshooting near peaks. |
Points outside the input grid are filled with left / right (both
default to numpy.nan). You can change these to 0.0 or any other
sentinel value if your downstream steps cannot handle NaN.
That is the core idea behind dynamic transformers: pipelines that stay self-contained and reusable even when correction parameters are not known until prediction time.
参考
XAxisInterpolator — full API reference.