Monitoreo de glucosa con PLS#
💡NOTA: Este documento es un notebook de Jupyter. ¡Puedes descargar el archivo fuente y ejecutarlo en tu entorno Jupyter!
Introducción#
Este conjunto de datos proporciona información sobre fermentación de etanol lignocelulósico a través de datos espectroscópicos recopilados usando espectroscopía de reflectancia total atenuada infrarroja media (ATR-MIR), junto con mediciones de referencia usando cromatografía líquida de alto rendimiento (HPLC) para validación.
El proyecto contiene dos conjuntos de datos:
Conjunto de datos de entrenamiento: Contiene datos espectrales con mediciones HPLC correspondientes usadas para entrenar los modelos de mínimos cuadrados parciales (PLS).
Conjunto de datos de prueba: Incluye una serie temporal de espectros recopilados durante la fermentación, más mediciones HPLC fuera de línea.
Para más información sobre estos conjuntos de datos y cómo pueden usarse para monitorear la fermentación, consulta nuestro artículo: «Transformando datos en información: Un modelo híbrido paralelo para estimación de estado en tiempo real en fermentación de etanol lignocelulósico». (Nota que los datos en el artículo difieren de los datos proporcionados aquí.)
Objetivo#
En este ejercicio, construirás un modelo PLS para monitorear la concentración de glucosa en tiempo real usando espectroscopía ATR-MIR. Entrenarás el modelo usando un pequeño conjunto de entrenamiento de espectros enriquecidos, luego probarás su rendimiento con espectros de un proceso de fermentación real.
Antes de comenzar#
Antes de comenzar, debes asegurarte de tener las siguientes dependencias instaladas:
chemotools
matplotlib
numpy
pandas
scikit-learn
Puedes instalarlas usando
pip install chemotools
pip install matplotlib
pip install numpy
pip install pandas
pip install scikit-learn
Cargando los datos de entrenamiento#
Puedes acceder desde el módulo chemotools.datasets con la función load_fermentation_train().
[ ]:
from chemotools.datasets import load_fermentation_train
spectra, hplc = load_fermentation_train()
La función load_fermentation_train() devuelve dos pandas.DataFrame:
spectra: Este dataframe contiene datos espectrales, con columnas que representan números de onda y filas que representan muestras.hplc: Aquí encontrarás mediciones HPLC de referencia para la concentración de glucosa (en g/L) de cada muestra, almacenadas en una sola columna etiquetada comoglucose.
💡NOTA: Si estás interesado en trabajar con
polars.DataFramesimplemente puedes usarload_fermentation_train(set_output="polars"). Nota que si eliges trabajar conpolars.DataFramelos números de onda se dan en los nombres de columna comostry no comofloat. Esto se debe a quepolarsno soporta nombres de columna con tipos distintos destr. Para extraer los números de onda comofloatdelpolars.DataFramepuedes usar el métododf.columns.to_numpy(dtype=np.float64).
Explorando los datos de entrenamiento#
Antes de comenzar con el modelado de datos, es importante familiarizarse con tus datos. Comencemos respondiendo algunas preguntas básicas:
¿Cuántas muestras hay?
¿y cuántos números de onda están disponibles?
[ ]:
print(f"Number of samples: {spectra.shape[0]}")
print(f"Number of wavenumbers: {spectra.shape[1]}")
Number of samples: 21
Number of wavenumbers: 1047
Ahora que tenemos lo básico, echemos un vistazo más de cerca a los datos.
Para los datos espectrales, puedes usar el método pandas.DataFrame.head() para examinar las primeras 5 filas:
[ ]:
spectra.head()
| 428.0 | 429.0 | 431.0 | 432.0 | 434.0 | 436.0 | 437.0 | 439.0 | 440.0 | 442.0 | ... | 1821.0 | 1823.0 | 1824.0 | 1825.0 | 1826.0 | 1828.0 | 1829.0 | 1830.0 | 1831.0 | 1833.0 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0.493506 | -0.383721 | 0.356846 | 0.205714 | 0.217082 | 0.740331 | 0.581749 | 0.106719 | 0.507973 | 0.298643 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 0.108225 | 0.488372 | -0.037344 | 0.448571 | 0.338078 | 0.632597 | 0.368821 | 0.462451 | 0.530752 | 0.221719 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 0.095238 | 0.790698 | 0.174274 | 0.314286 | 0.106762 | 0.560773 | 0.182510 | 0.482213 | 0.482916 | 0.341629 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | 0.666667 | 0.197674 | 0.352697 | 0.385714 | 0.405694 | 0.508287 | 0.463878 | 0.430830 | 0.455581 | 0.527149 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | 0.532468 | -0.255814 | 0.078838 | 0.057143 | 0.238434 | 0.997238 | 0.399240 | 0.201581 | 0.533030 | 0.246606 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
5 rows × 1047 columns
Por brevedad, no mostraremos la tabla completa aquí, pero notarás que cada columna corresponde a un número de onda, y cada fila representa una muestra.
En cuanto a los datos HPLC, el método pandas.DataFrame.describe() proporciona un resumen de la columna de glucosa:
[ ]:
hplc.describe()
| glucose | |
|---|---|
| count | 21.000000 |
| mean | 19.063895 |
| std | 12.431570 |
| min | 0.000000 |
| 25% | 9.057189 |
| 50% | 18.395220 |
| 75% | 29.135105 |
| max | 38.053004 |
Este resumen ofrece información sobre la distribución de la concentración de glucosa . Con este conocimiento práctico, ahora estamos listos para continuar con nuestro viaje de modelado de datos.
Visualizando los datos de entrenamiento#
Para entender mejor nuestro conjunto de datos, visualizaremos los datos. Graficaremos los espectros en el conjunto de datos de entrenamiento, con cada espectro codificado por color para reflejar su concentración de glucosa asociada. Este enfoque visual proporciona un medio didáctico para entender las características del conjunto de datos, ofreciendo información sobre variaciones químicas entre las muestras. Para hacerlo, usaremos el módulo matplotlib.pyplot. Recuerda instalarlo primero usando:
pip install matplotlib
Hasta ahora, hemos usado pandas.DataFrame para representar el conjunto de datos. Los pandas.DataFrame son excelentes para almacenar y manipular muchos conjuntos de datos grandes. Sin embargo, a menudo encuentro más conveniente usar numpy.ndarray para trabajar con datos espectrales. Por lo tanto, convertiremos el pandas.DataFrame a numpy.ndarray usando el método pandas.DataFrame.to_numpy().
Así que nuestro primer paso será transformar nuestro pandas.DataFrame a numpy.ndarray:
[ ]:
import numpy as np
# Convert the spectra pandas.DataFrame to numpy.ndarray
spectra_np = spectra.to_numpy()
# Convert the wavenumbers pandas.columns to numpy.ndarray
wavenumbers = spectra.columns.to_numpy(dtype=np.float64)
# Convert the hplc pandas.DataFrame to numpy.ndarray
hplc_np = hplc.to_numpy()
Antes de graficar nuestros datos, crearemos una función de graficación dedicada que codifique por color cada espectro según su concentración de glucosa. Este enfoque ayuda a visualizar la relación entre patrones espectrales y niveles de glucosa.
La función implementará:
Normalización de color: Usaremos
matplotlib.colors.Normalizepara escalar valores de concentración de glucosa a un rango 0-1, haciéndolos adecuados para mapeo de color.Mapeo de color: Los valores normalizados se mapearán a colores usando un mapa de colores vía
matplotlib.cm.ScalarMappable.Barra de color: Agregaremos una barra de color para indicar cómo los colores corresponden a valores de concentración de glucosa.
Esta visualización nos permite comparar múltiples espectros basados en sus concentraciones de glucosa asociadas.
[ ]:
import matplotlib.pyplot as plt
import numpy as np
from matplotlib.colors import Normalize
def plot_spectra(
spectra: np.ndarray, wavenumbers: np.ndarray, hplc: np.ndarray, zoom_in=False
):
"""
Plot spectra with colors based on glucose concentration.
Parameters:
-----------
spectra : np.ndarray
Array containing spectral data, where each row is a spectrum
wavenumbers : np.ndarray
Array of wavenumbers (x-axis values)
hplc : np.ndarray
Array of glucose concentrations corresponding to each spectrum
zoom_in : bool, default=False
If True, zooms into a specific region of interest (950-1550 cm⁻¹)
Returns:
--------
None
Displays the plot
"""
# Create figure and axis
fig, ax = plt.subplots(figsize=(10, 3))
# Set up color mapping
cmap = plt.get_cmap("cool") # Cool colormap (purple to blue)
normalize = Normalize(vmin=hplc.min(), vmax=hplc.max())
# Plot each spectrum with appropriate color
for i, spectrum in enumerate(spectra):
color = cmap(normalize(hplc[i]))
ax.plot(wavenumbers, spectrum, color=color, linewidth=1.5, alpha=0.8)
# Add colorbar to show glucose concentration scale
sm = plt.cm.ScalarMappable(cmap=cmap, norm=normalize)
sm.set_array([])
fig.colorbar(sm, ax=ax, label="Glucose (g/L)")
# Set axis labels and title
ax.set_xlabel("Wavenumber (cm$^{-1}$)")
ax.set_ylabel("Absorbance (a.u.)")
ax.set_title("Fermentation Training Set Spectra")
# Apply zoom if requested
if zoom_in:
ax.set_xlim(950, 1550)
ax.set_ylim(0.45, 0.65)
ax.set_title("Fermentation Training Set Spectra (Zoomed Region)")
# Improve appearance
ax.grid(True, linestyle="--", alpha=0.6)
fig.tight_layout()
# Show the plot
plt.show()
Ahora podemos usar esta función para graficar el conjunto de datos de entrenamiento:
[ ]:
plot_spectra(spectra_np, wavenumbers, hplc_np)
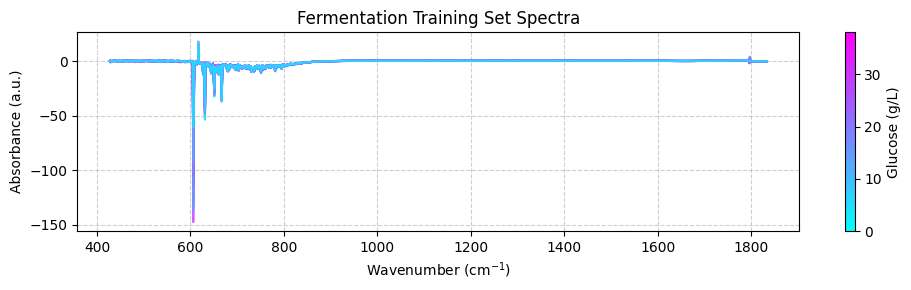
Estos espectros pueden verse poco claros porque cubren un amplio rango de números de onda que incluye áreas con poca información química. Para obtener una mejor vista, debemos enfocarnos en la región «huella digital» entre 950 y 1550 cm-1, donde aparecen las características químicas importantes. Por ahora, usaremos el parámetro zoom_in=True en nuestra función plot_spectra() para enfocarnos en esta región. Más tarde, aprenderemos cómo recortar apropiadamente los espectros usando funciones de preprocesamiento.
[ ]:
plot_spectra(spectra_np, wavenumbers, hplc_np, zoom_in=True)
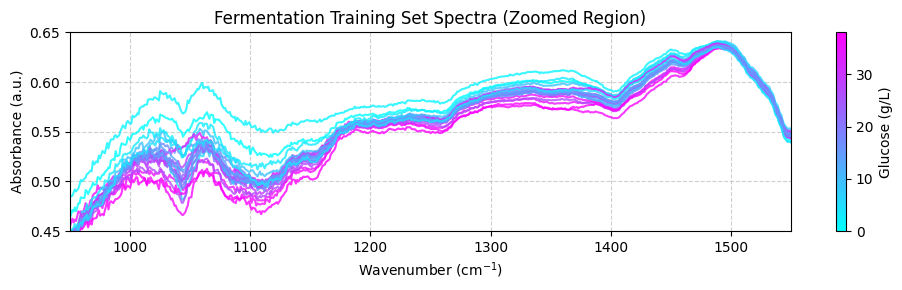
Preprocesando los datos de entrenamiento#
Ahora que has explorado el conjunto de datos, es hora de preprocesar los datos espectrales. Este paso es esencial para eliminar variaciones no deseadas, como desplazamientos de línea base y ruido, que pueden impactar negativamente el rendimiento del modelo. Usaremos los módulos chemotools y scikit-learn para preprocesar los datos espectrales.
Preprocesaremos los espectros usando los siguientes pasos:
Corte de rango: para eliminar los números de onda fuera del rango entre 950 y 1550 cm-1.
Corrección lineal: para eliminar el desplazamiento de línea base lineal.
Savitzky-Golay: calcula la derivada de n-ésimo orden de los espectros usando el método de Savitzky-Golay. Esto es útil para eliminar efectos de dispersión aditivos y multiplicativos.
Escalador estándar: para escalar los espectros a media cero.
Encadenaremos los pasos de preprocesamiento usando la función `make_pipeline() <https://scikit-learn.org/stable/modules/generated/sklearn.pipeline.make_pipeline.html>`__ de scikit-learn.
💡 NOTA: ¿Qué es un pipeline? Un pipeline es una secuencia de pasos que se ejecutan en un orden específico. En nuestro caso, crearemos un pipeline que ejecutará los pasos de preprocesamiento en el orden descrito arriba. Puedes encontrar más información sobre cómo trabajar con pipelines en nuestra página de documentación.
[ ]:
from sklearn.pipeline import make_pipeline
from sklearn.preprocessing import StandardScaler
from chemotools.baseline import LinearCorrection
from chemotools.derivative import SavitzkyGolay
from chemotools.feature_selection import RangeCut
# create a pipeline that scales the data
preprocessing = make_pipeline(
RangeCut(start=950, end=1500, wavenumbers=wavenumbers),
LinearCorrection(),
SavitzkyGolay(window_size=15, polynomial_order=2, derivate_order=1),
StandardScaler(with_std=False),
)
Ahora podemos usar el pipeline de preprocesamiento para preprocesar los espectros:
[ ]:
spectra_preprocessed = preprocessing.fit_transform(spectra_np)
Y podemos usar la función plot_spectra() para graficar los espectros preprocesados:
[ ]:
# get the wavenumbers after the range cut
wavenumbers_cut = preprocessing.named_steps["rangecut"].wavenumbers_
# plot the preprocessed spectra
plot_spectra(spectra_preprocessed, wavenumbers_cut, hplc_np)
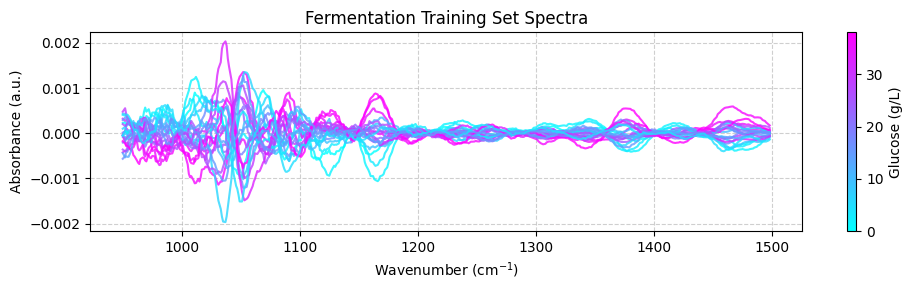
💡 NOTA: ¡Esto es genial! ¿Ves cómo estamos integrando quimiometría con scikit-learn?
RangeCut,LinearCorrectionySavitizkyGolayson todas técnicas de preprocesamiento implementadas enchemotoolsmientras queStandardScalerypipelinesson funcionalidades proporcionadas porscikit-learn. Este es el poder dechemotools— está diseñado para trabajar sin problemas conscikit-learn
Entrenando un modelo PLS#
Los Mínimos Cuadrados Parciales (PLS) son una técnica de regresión poderosa comúnmente usada en quimiometría. Funciona encontrando una representación de espacio latente que maximiza la covarianza con la variable objetivo.
Un parámetro crítico en el modelado PLS es el número de componentes (dimensiones) en el espacio latente:
Demasiados componentes lleva al sobreajuste, donde el modelo captura ruido en lugar de señal
Muy pocos componentes resulta en subajuste, perdiendo patrones importantes en los datos
Al trabajar con muestras limitadas, la validación cruzada proporciona un método confiable para seleccionar el número óptimo de componentes. Este enfoque:
Divide los datos en conjuntos de entrenamiento y validación
Entrena el modelo en el conjunto de entrenamiento y lo evalúa en el conjunto de validación
Repite este proceso con diferentes divisiones de datos
Promedia los resultados para estimar el error de generalización del modelo
Para nuestro análisis, usaremos la clase GridSearchCV de scikit-learn para probar sistemáticamente diferentes números de componentes mediante validación cruzada. Esto identificará la complejidad óptima para nuestro modelo PLS basándose en métricas de rendimiento a través de múltiples divisiones de datos.
[ ]:
# import the PLSRegression and GridSearchCV classes
from sklearn.cross_decomposition import PLSRegression
from sklearn.model_selection import GridSearchCV
# instanciate a PLSRegression object
pls = PLSRegression(scale=False)
# define the parameter grid (number of components to evaluate)
param_grid = {"n_components": [1, 2, 3, 4, 5, 6, 7, 8, 9, 10]}
# create a grid search object
grid_search = GridSearchCV(pls, param_grid, cv=10, scoring="neg_mean_absolute_error")
# fit the grid search object to the data
grid_search.fit(spectra_preprocessed, hplc_np)
# print the best parameters and score
print("Best parameters: ", grid_search.best_params_)
print("Best score: ", np.abs(grid_search.best_score_))
Best parameters: {'n_components': 6}
Best score: 0.922944026246299
Sugiriendo que el número óptimo de componentes es 6, con un error absoluto medio de 0.92 g/L. Podemos verificar esto graficando el error absoluto medio en función del número de componentes:
[ ]:
fig, ax = plt.subplots(figsize=(10, 3))
ax.plot(
param_grid["n_components"],
np.abs(grid_search.cv_results_["mean_test_score"]),
marker="o",
color="b",
)
ax.set_xlabel("Number of components")
ax.set_ylabel("Mean absolute error (g/L)")
ax.set_title("Cross validation results")
ax.grid(True, linestyle="--", alpha=0.6)
fig.tight_layout()
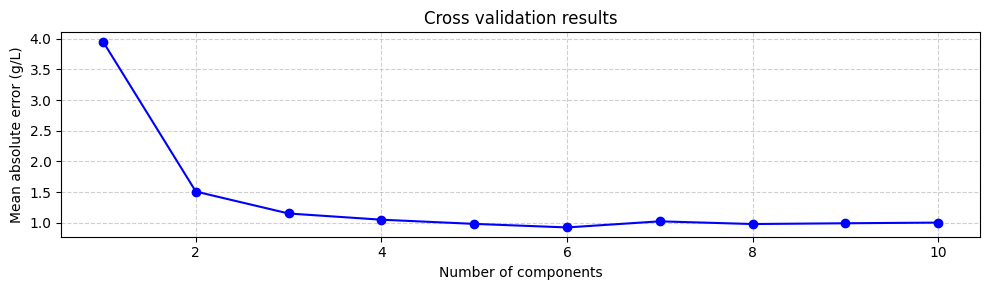
Aunque usar el número de componentes que minimiza el error absoluto medio es un buen punto de partida, no siempre es lo mejor. El modelo con 6 componentes no aumenta mucho el error absoluto medio comparado con el modelo con 3 o incluso dos componentes. Sin embargo, el modelo con 6 componentes incluye componentes asociados con valores propios pequeños, que son más inciertos. Esto significa que modelos con 3 o 2 componentes podrían ser más robustos. Por lo tanto, siempre es una buena idea probar diferentes números de componentes y seleccionar el que dé el mejor rendimiento. Sin embargo, por ahora entrenaremos el modelo con 6 componentes:
[ ]:
# instanciate a PLSRegression object with 6 components
pls = PLSRegression(n_components=6, scale=False)
# fit the model to the data
pls.fit(spectra_preprocessed, hplc_np)
PLSRegression(n_components=6, scale=False)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook.
On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
PLSRegression(n_components=6, scale=False)
Finalmente podemos evaluar el rendimiento del modelo en el conjunto de entrenamiento:
[ ]:
# predict the glucose concentrations
hplc_pred = pls.predict(spectra_preprocessed)
# plot the predictions
fig, ax = plt.subplots(figsize=(4, 4))
ax.scatter(hplc_np, hplc_pred, color="blue")
ax.plot([0, 40], [0, 40], color="magenta")
ax.set_xlabel("Reference glucose (g/L)")
ax.set_ylabel("Predicted glucose (g/L)")
ax.set_title("PLS regression")
ax.grid(True, linestyle="--", alpha=0.6)
fig.tight_layout()
plt.show()
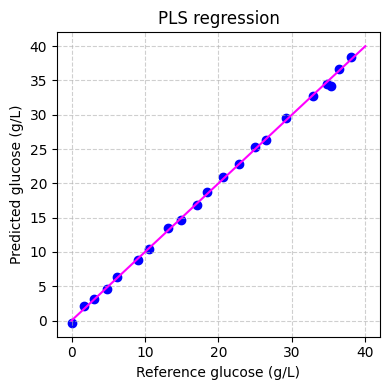
Aplicando el modelo#
Ahora que hemos entrenado nuestro modelo, podemos aplicarlo al conjunto de datos de prueba . El conjunto de datos de prueba contiene espectros registrados en tiempo real durante el proceso de fermentación. El conjunto de datos de prueba contiene dos pandas.DataFrame:
spectra: Este conjunto de datos contiene datos espectrales, con columnas que representan números de onda y filas que representan muestras. Estos espectros fueron registrados en tiempo real durante el proceso de fermentación aproximadamente cada 1.5 minutos.hplc: Este conjunto de datos contiene mediciones HPLC, específicamente concentraciones de glucosa (en g/L), almacenadas en una sola columna etiquetada comoglucose. Estas mediciones fueron registradas fuera de línea aproximadamente cada 60 minutos.
Usaremos la función load_fermentation_test() del módulo chemotools.datasets para cargar el conjunto de datos de prueba:
[ ]:
from chemotools.datasets import load_fermentation_test
spectra_test, hplc_test = load_fermentation_test()
Luego, preprocesaremos los espectros usando el mismo pipeline de preprocesamiento que usamos para el conjunto de datos de entrenamiento:
[ ]:
# convert the spectra pandas.DataFrame to numpy.ndarray
spectra_test_np = spectra_test.to_numpy()
# preprocess the spectra
spectra_test_preprocessed = preprocessing.transform(spectra_test_np)
Finalmente, podemos usar el modelo PLS para predecir las concentraciones de glucosa:
[ ]:
# predict the glucose concentrations
glucose_test_pred = pls.predict(spectra_test_preprocessed)
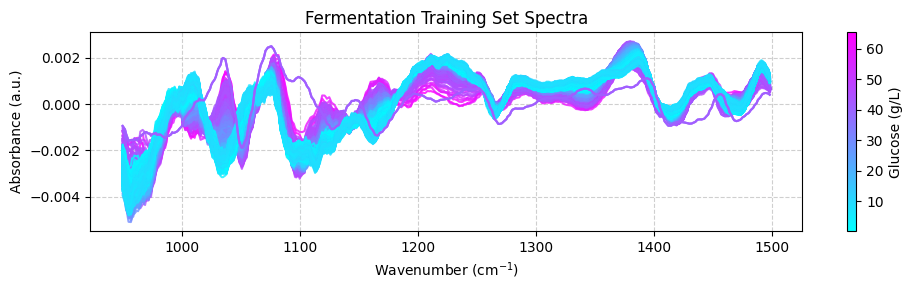
Podemos usar los valores predichos para graficar los espectros codificados por color según las concentraciones de glucosa predichas:
[ ]:
plot_spectra(spectra_test_preprocessed, wavenumbers_cut, glucose_test_pred)
Ahora podemos comparar las concentraciones de glucosa predichas con las mediciones HPLC fuera de línea. Graficaremos las predicciones y las mediciones fuera de línea a lo largo del tiempo. Cada espectro fue tomado cada 1.25 minutos, mientras que las mediciones fuera de línea fueron tomadas cada 60 minutos.
[ ]:
# make linspace of length of predictoins
time = (
np.linspace(
0,
len(glucose_test_pred),
len(glucose_test_pred),
)
* 1.25
/ 60
)
# plot the predictions
fig, ax = plt.subplots(figsize=(10, 3))
ax.plot(time, glucose_test_pred, color="blue", label="Predicted")
ax.plot(
hplc_test.index, hplc_test["glucose"] + 4, "o", color="fuchsia", label="Measured"
)
ax.set_xlabel("Time (h)")
ax.set_ylabel("Glucose (g/L)")
ax.set_title("Fermentation test set")
ax.legend()
ax.grid(True, linestyle="--", alpha=0.6)
fig.tight_layout()
plt.show()
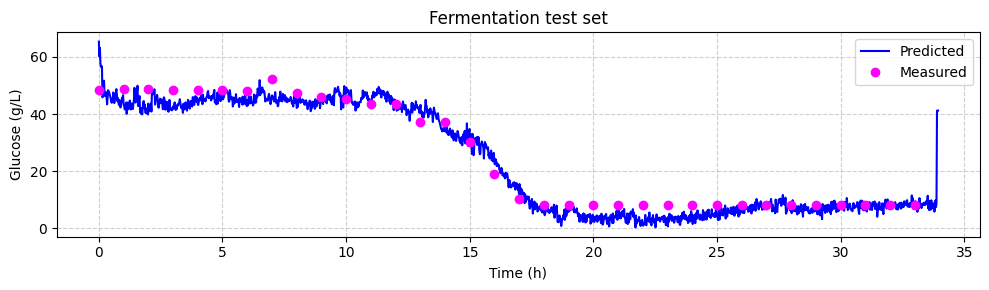
Resumen#
En este tutorial, exploramos cómo aplicar aprendizaje automático al análisis de datos espectroscópicos usando el conjunto de datos de Fermentación. Nuestro objetivo era construir un modelo de regresión capaz de predecir concentraciones de glucosa durante la fermentación de etanol lignocelulósico. Aquí hay un resumen rápido de lo que cubrimos:
Introducción: Comenzamos introduciendo el conjunto de datos de Fermentación, que incluye datos espectrales recopilados vía espectroscopía infrarroja media de reflectancia total atenuada (ATR-MIR) y datos de referencia HPLC correspondientes. Este conjunto de datos es especialmente valioso para estudiar procesos de fermentación en tiempo real.
Cargando y explorando los datos: Cargamos los datos de entrenamiento, examinamos su estructura y echamos un vistazo más de cerca a las mediciones espectrales y HPLC . Entender los datos en esta etapa es una base crítica para cualquier análisis exitoso.
Visualización de datos: A través de la visualización, obtuvimos una imagen más clara del conjunto de datos. Al graficar espectros codificados por color según la concentración de glucosa , pudimos observar cómo aparecen las variaciones químicas en las muestras.
Preprocesamiento de datos: Preparamos los datos espectrales para el modelado aplicando varias técnicas de preprocesamiento: corte de rango, corrección lineal , derivadas de Savitzky-Golay y escalado estándar. Estos pasos ayudaron a reducir el ruido y mejorar la calidad general de los datos.
Entrenamiento del modelo: Usando los datos procesados, entrenamos un modelo de regresión de Mínimos Cuadrados Parciales (PLS) para predecir concentraciones de glucosa. La validación cruzada nos ayudó a determinar el número óptimo de componentes y evaluar el rendimiento del modelo.
Prueba y aplicación: Finalmente, aplicamos nuestro modelo entrenado al conjunto de datos de prueba para predecir concentraciones de glucosa en tiempo real. Esto demostró cómo el modelo puede usarse para rastrear niveles de glucosa durante la fermentación.
Este tutorial ofrece una introducción práctica al uso de aprendizaje automático para análisis de datos espectroscópicos. Siguiendo estos pasos, puedes extraer información significativa de datos espectrales complejos y construir modelos predictivos que tienen aplicaciones en el mundo real.