PLSによるグルコースモニタリング#
💡 注意:このドキュメントはJupyterノートブックです。ソースファイルをダウンロードして、Jupyter環境で実行できます!
はじめに#
このデータセットは、減衰全反射中赤外(ATR-MIR)分光法を使用して収集された分光データを通じて、リグノセルロース系エタノール発酵に関する情報を提供します。検証には高速液体クロマトグラフィー(HPLC)を使用した参照測定も含まれています。
プロジェクトには2つのデータセットが含まれています:
訓練データセット:部分最小二乗(PLS)モデルを訓練するために使用される、対応するHPLC測定値を含むスペクトルデータが含まれています。
テストデータセット:発酵中に収集されたスペクトルの時系列と、オフラインHPLC測定値が含まれています。
これらのデータセットと発酵のモニタリングへの使用方法の詳細については、論文「Transforming Data to Information: A Parallel Hybrid Model for Real-Time State Estimation in Lignocellulosic Ethanol Fermentation」を参照してください。(論文のデータとここで提供されるデータは異なることに注意してください。)
目的#
この演習では、ATR-MIR分光法を使用してグルコース濃度をリアルタイムでモニタリングするPLSモデルを構築します。スパイクされたスペクトルの小さな訓練セットを使用してモデルを訓練し、実際の発酵プロセスからのスペクトルでパフォーマンスをテストします。
始める前に#
始める前に、以下の依存関係がインストールされていることを確認する必要があります:
chemotools
matplotlib
numpy
pandas
scikit-learn
次のコマンドでインストールできます
pip install chemotools
pip install matplotlib
pip install numpy
pip install pandas
pip install scikit-learn
訓練データのロード#
chemotools.datasets モジュールの load_fermentation_train() 関数からアクセスできます。
[ ]:
from chemotools.datasets import load_fermentation_train
spectra, hplc = load_fermentation_train()
load_fermentation_train() 関数は2つの pandas.DataFrame を返します:
spectra:このデータフレームにはスペクトルデータが含まれており、列は波数を表し、行はサンプルを表します。hplc:ここでは、各サンプルのグルコース濃度(g/L)の参照HPLC測定値が、glucoseというラベルの単一列に格納されています。
💡 注意:
polars.DataFrameを使用する場合は、load_fermentation_train(set_output="polars")を使用できます。polars.DataFrameを使用する場合、波数はfloatではなくstrとして列名に指定されることに注意してください。これは、polarsがstr以外の型の列名をサポートしていないためです。polars.DataFrameからfloatとして波数を抽出するには、df.columns.to_numpy(dtype=np.float64)メソッドを使用できます。
訓練データの探索#
データモデリングを開始する前に、データに慣れることが重要です。いくつかの基本的な質問に答えることから始めましょう:
サンプルはいくつありますか?
利用可能な波数はいくつですか?
[ ]:
print(f"Number of samples: {spectra.shape[0]}")
print(f"Number of wavenumbers: {spectra.shape[1]}")
Number of samples: 21
Number of wavenumbers: 1047
基本を理解したので、データをより詳しく見てみましょう。
スペクトルデータの場合、 pandas.DataFrame.head() メソッドを使用して最初の5行を調べることができます:
[ ]:
spectra.head()
| 428.0 | 429.0 | 431.0 | 432.0 | 434.0 | 436.0 | 437.0 | 439.0 | 440.0 | 442.0 | ... | 1821.0 | 1823.0 | 1824.0 | 1825.0 | 1826.0 | 1828.0 | 1829.0 | 1830.0 | 1831.0 | 1833.0 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0.493506 | -0.383721 | 0.356846 | 0.205714 | 0.217082 | 0.740331 | 0.581749 | 0.106719 | 0.507973 | 0.298643 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 0.108225 | 0.488372 | -0.037344 | 0.448571 | 0.338078 | 0.632597 | 0.368821 | 0.462451 | 0.530752 | 0.221719 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 0.095238 | 0.790698 | 0.174274 | 0.314286 | 0.106762 | 0.560773 | 0.182510 | 0.482213 | 0.482916 | 0.341629 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | 0.666667 | 0.197674 | 0.352697 | 0.385714 | 0.405694 | 0.508287 | 0.463878 | 0.430830 | 0.455581 | 0.527149 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | 0.532468 | -0.255814 | 0.078838 | 0.057143 | 0.238434 | 0.997238 | 0.399240 | 0.201581 | 0.533030 | 0.246606 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
5 rows × 1047 columns
簡潔にするために、ここでは全テーブルを表示しませんが、各列が波数に対応し、各行がサンプルを表していることに気づくでしょう。
HPLCデータに移ると、 pandas.DataFrame.describe() メソッドがグルコース列の要約を提供します:
[ ]:
hplc.describe()
| glucose | |
|---|---|
| count | 21.000000 |
| mean | 19.063895 |
| std | 12.431570 |
| min | 0.000000 |
| 25% | 9.057189 |
| 50% | 18.395220 |
| 75% | 29.135105 |
| max | 38.053004 |
この要約は、グルコース濃度の分布に関する洞察を提供します。この実用的な知識により、データモデリングの旅を続ける準備が整いました。
訓練データの視覚化#
データセットをより よく理解するために、データを視覚化します。訓練データセット内のスペクトルをプロットし、各スペクトルを関連するグルコース濃度を反映する色でコーディングします。この視覚的なアプローチは、データセットの特性を理解するための教育的な手段を提供し、サンプル間の化学的変動に関する洞察を提供します。これを行うには、 matplotlib.pyplot モジュールを使用します。最初に次のコマンドでインストールすることを忘れないでください:
pip install matplotlib
これまで、データセットを表すために pandas.DataFrame を使用してきました。 pandas.DataFrame は、多くの大規模データセットを保存および操作するのに優れています。ただし、スペクトルデータを扱う場合は numpy.ndarray を使用する方が便利だと感じることがよくあります。したがって、 pandas.DataFrame.to_numpy() メソッドを使用して pandas.DataFrame を numpy.ndarray に変換します。
最初のステップは、 pandas.DataFrame を numpy.ndarray に変換することです:
[ ]:
import numpy as np
# Convert the spectra pandas.DataFrame to numpy.ndarray
spectra_np = spectra.to_numpy()
# Convert the wavenumbers pandas.columns to numpy.ndarray
wavenumbers = spectra.columns.to_numpy(dtype=np.float64)
# Convert the hplc pandas.DataFrame to numpy.ndarray
hplc_np = hplc.to_numpy()
データをプロットする前に、各スペクトルをグルコース濃度に応じて色分けする専用のプロット関数を作成します。このアプローチは、スペクトルパターンとグルコースレベルの関係を視覚化するのに役立ちます。
関数は次を実装します:
色の正規化:
matplotlib.colors.Normalizeを使用してグルコース濃度値を0-1の範囲にスケーリングし、色マッピングに適したものにします。色マッピング:正規化された値は、
matplotlib.cm.ScalarMappableを介してカラーマップを使用して色にマッピングされます。カラーバー:色がグルコース濃度値にどのように対応するかを示すカラーバーを追加します。
この視覚化により、関連するグルコース濃度に基づいて複数のスペクトルを比較できます。
[ ]:
import matplotlib.pyplot as plt
import numpy as np
from matplotlib.colors import Normalize
def plot_spectra(
spectra: np.ndarray, wavenumbers: np.ndarray, hplc: np.ndarray, zoom_in=False
):
"""
Plot spectra with colors based on glucose concentration.
Parameters:
-----------
spectra : np.ndarray
Array containing spectral data, where each row is a spectrum
wavenumbers : np.ndarray
Array of wavenumbers (x-axis values)
hplc : np.ndarray
Array of glucose concentrations corresponding to each spectrum
zoom_in : bool, default=False
If True, zooms into a specific region of interest (950-1550 cm⁻¹)
Returns:
--------
None
Displays the plot
"""
# Create figure and axis
fig, ax = plt.subplots(figsize=(10, 3))
# Set up color mapping
cmap = plt.get_cmap("cool") # Cool colormap (purple to blue)
normalize = Normalize(vmin=hplc.min(), vmax=hplc.max())
# Plot each spectrum with appropriate color
for i, spectrum in enumerate(spectra):
color = cmap(normalize(hplc[i]))
ax.plot(wavenumbers, spectrum, color=color, linewidth=1.5, alpha=0.8)
# Add colorbar to show glucose concentration scale
sm = plt.cm.ScalarMappable(cmap=cmap, norm=normalize)
sm.set_array([])
fig.colorbar(sm, ax=ax, label="Glucose (g/L)")
# Set axis labels and title
ax.set_xlabel("Wavenumber (cm$^{-1}$)")
ax.set_ylabel("Absorbance (a.u.)")
ax.set_title("Fermentation Training Set Spectra")
# Apply zoom if requested
if zoom_in:
ax.set_xlim(950, 1550)
ax.set_ylim(0.45, 0.65)
ax.set_title("Fermentation Training Set Spectra (Zoomed Region)")
# Improve appearance
ax.grid(True, linestyle="--", alpha=0.6)
fig.tight_layout()
# Show the plot
plt.show()
この関数を使用して訓練データセットをプロットできます:
[ ]:
plot_spectra(spectra_np, wavenumbers, hplc_np)
これらのスペクトルは不明瞭に見えるかもしれません。これは、化学情報がほとんどない領域を含む広い波数範囲をカバーしているためです。より良い表示を得るには、重要な化学的特徴が現れる950から1550 cm-1の間の「指紋」領域に焦点を当てる必要があります。今のところ、 plot_spectra() 関数の zoom_in=True パラメータを使用してこの領域に焦点を当てます。後で、前処理関数を使用してスペクトルを適切にトリミングする方法を学びます。
[ ]:
plot_spectra(spectra_np, wavenumbers, hplc_np, zoom_in=True)
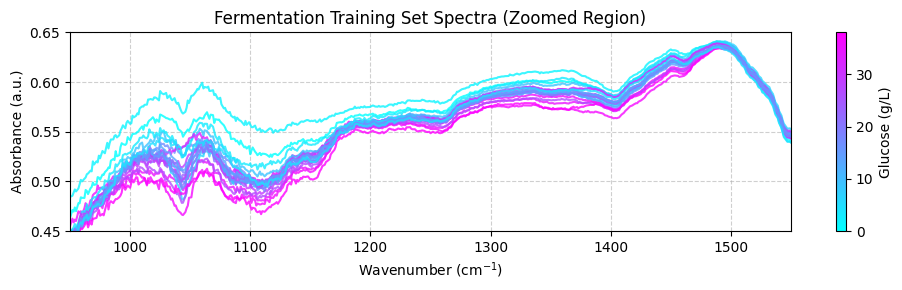
訓練データの前処理#
データセットを探索したので、スペクトルデータを前処理する時が来ました。このステップは、ベースラインシフトやノイズなどの不要な変動を除去するために不可欠です。これらは、モデルのパフォーマンスに悪影響を与える可能性があります。 chemotools と scikit-learn モジュールを使用してスペクトルデータを前処理します。
次のステップを使用してスペクトルを前処理します:
Range Cut :950から1550 cm-1の範囲外の波数を除去します。
Linear Correction :線形ベースラインシフトを除去します。
Savitzky-Golay :Savitzky-Golay法を使用してスペクトルのn次導関数を計算します。これは、加法および乗法の散乱効果を除去するのに役立ちます。
Standard Scaler :スペクトルをゼロ平均にスケーリングします。
scikit-learn の make_pipeline() 詳細については〜を参照してください 関数を使用して前処理ステップを連鎖させます。
💡 注意:パイプラインとは?パイプラインは、特定の順序で実行されるステップのシーケンスです。今回の場合、上記の順序で前処理ステップを実行するパイプラインを作成します。パイプラインの使用方法の詳細については、 ドキュメントページ を参照してください。
[ ]:
from sklearn.pipeline import make_pipeline
from sklearn.preprocessing import StandardScaler
from chemotools.baseline import LinearCorrection
from chemotools.derivative import SavitzkyGolay
from chemotools.feature_selection import RangeCut
# create a pipeline that scales the data
preprocessing = make_pipeline(
RangeCut(start=950, end=1500, wavenumbers=wavenumbers),
LinearCorrection(),
SavitzkyGolay(window_size=15, polynomial_order=2, derivate_order=1),
StandardScaler(with_std=False),
)
前処理パイプラインを使用してスペクトルを前処理できます:
[ ]:
spectra_preprocessed = preprocessing.fit_transform(spectra_np)
そして、 plot_spectra() 関数を使用して前処理されたスペクトルをプロットできます:
[ ]:
# get the wavenumbers after the range cut
wavenumbers_cut = preprocessing.named_steps["rangecut"].wavenumbers_
# plot the preprocessed spectra
plot_spectra(spectra_preprocessed, wavenumbers_cut, hplc_np)
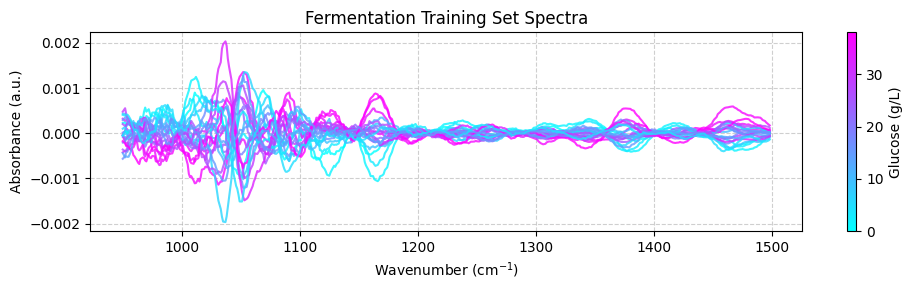
💡 注意:これは素晴らしいです!ケモメトリクスを
scikit-learnとどのように統合しているかご覧ください。RangeCut、LinearCorrection、SavitizkyGolayはすべてchemotoolsに実装された前処理技術であり、StandardScalerとpipelinesはscikit-learnによって提供される機能です。これがchemotoolsの力であり、scikit-learnとシームレスに動作するように設計されています。
PLSモデルの訓練#
部分最小二乗(PLS)は、ケモメトリクスで一般的に使用される強力な回帰技術です。目的変数との共分散を最大化する潜在空間表現を見つけることで機能します。
PLSモデリングの重要なパラメータは、潜在空間のコンポーネント数(次元数)です:
コンポーネントが多すぎると過学習につながり、モデルがシグナルではなくノイズを捕捉します
コンポーネントが少なすぎると過小適合になり、データ内の重要なパターンを見逃します
限られたサンプルを扱う場合、交差検証は最適なコンポーネント数を選択するための信頼できる方法を提供します。このアプローチは:
データを訓練セットと検証セットに分割します
訓練セットでモデルを訓練し、検証セットで評価します
異なるデータ分割でこのプロセスを繰り返します
結果を平均してモデルの汎化誤差を推定します
分析には、scikit-learn の GridSearchCV クラスを使用して、交差検証を通じて異なるコンポーネント数を体系的にテストします。これにより、複数のデータ分割にわたるパフォーマンス指標に基づいてPLSモデルの最適な複雑さが特定されます。
[ ]:
# import the PLSRegression and GridSearchCV classes
from sklearn.cross_decomposition import PLSRegression
from sklearn.model_selection import GridSearchCV
# instanciate a PLSRegression object
pls = PLSRegression(scale=False)
# define the parameter grid (number of components to evaluate)
param_grid = {"n_components": [1, 2, 3, 4, 5, 6, 7, 8, 9, 10]}
# create a grid search object
grid_search = GridSearchCV(pls, param_grid, cv=10, scoring="neg_mean_absolute_error")
# fit the grid search object to the data
grid_search.fit(spectra_preprocessed, hplc_np)
# print the best parameters and score
print("Best parameters: ", grid_search.best_params_)
print("Best score: ", np.abs(grid_search.best_score_))
Best parameters: {'n_components': 6}
Best score: 0.922944026246299
最適なコンポーネント数は6で、平均絶対誤差は0.92 g/Lであることが示唆されています。これを検証するために、コンポーネント数の関数として平均絶対誤差をプロットできます:
[ ]:
fig, ax = plt.subplots(figsize=(10, 3))
ax.plot(
param_grid["n_components"],
np.abs(grid_search.cv_results_["mean_test_score"]),
marker="o",
color="b",
)
ax.set_xlabel("Number of components")
ax.set_ylabel("Mean absolute error (g/L)")
ax.set_title("Cross validation results")
ax.grid(True, linestyle="--", alpha=0.6)
fig.tight_layout()
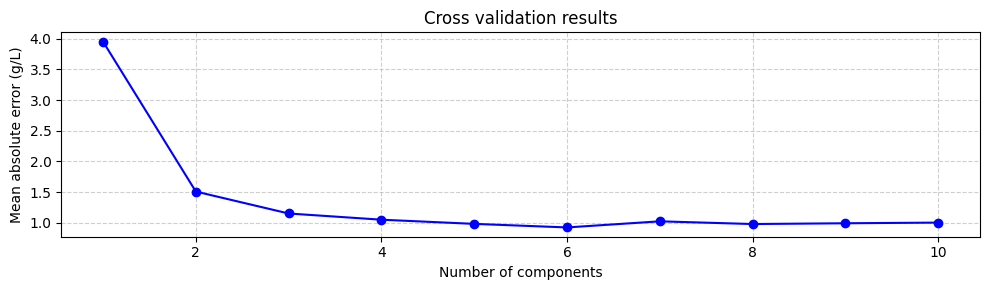
平均絶対誤差を最小化するコンポーネント数を使用することは良い出発点ですが、常に最良とは限りません。6コンポーネントモデルは、3コンポーネントまたは2コンポーネントモデルと比較して平均絶対誤差をあまり増加させません。しかし、6コンポーネントモデルには小さな固有値に関連するコンポーネントが含まれており、これらはより不確実です。これは、3または2コンポーネントのモデルの方がより堅牢である可能性があることを意味します。したがって、異なるコンポーネント数を試して、最高のパフォーマンスを提供するものを選択することは常に良い考えです。ただし、今のところ6コンポーネントでモデルを訓練します:
[ ]:
# instanciate a PLSRegression object with 6 components
pls = PLSRegression(n_components=6, scale=False)
# fit the model to the data
pls.fit(spectra_preprocessed, hplc_np)
PLSRegression(n_components=6, scale=False)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook.
On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
PLSRegression(n_components=6, scale=False)
最後に、訓練セットでモデルのパフォーマンスを評価できます:
[ ]:
# predict the glucose concentrations
hplc_pred = pls.predict(spectra_preprocessed)
# plot the predictions
fig, ax = plt.subplots(figsize=(4, 4))
ax.scatter(hplc_np, hplc_pred, color="blue")
ax.plot([0, 40], [0, 40], color="magenta")
ax.set_xlabel("Reference glucose (g/L)")
ax.set_ylabel("Predicted glucose (g/L)")
ax.set_title("PLS regression")
ax.grid(True, linestyle="--", alpha=0.6)
fig.tight_layout()
plt.show()
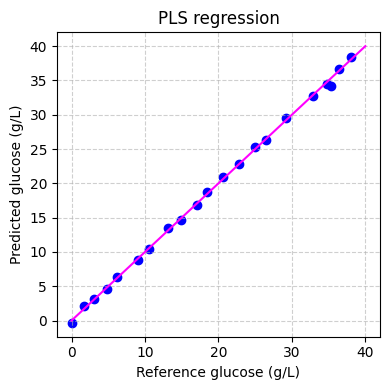
モデルの適用#
モデルを訓練したので、テストデータセットに適用できます。テストデータセットには、発酵プロセス中にリアルタイムで記録されたスペクトルが含まれています。テストデータセットには2つの pandas.DataFrame が含まれています:
spectra:このデータセットにはスペクトルデータが含まれており、列は波数を表し、行はサンプルを表します。これらのスペクトルは、発酵プロセス中に約1.5分ごとにリアルタイムで記録されました。hplc:このデータセットにはHPLC測定値、具体的にはグルコース濃度(g/L)が含まれており、glucoseというラベルの単一列に格納されています。これらの測定値は、約60分ごとにオフラインで記録されました。
chemotools.datasets モジュールの load_fermentation_test() 関数を使用してテストデータセットをロードします:
[ ]:
from chemotools.datasets import load_fermentation_test
spectra_test, hplc_test = load_fermentation_test()
次に、訓練データセットに使用したのと同じ前処理パイプラインを使用してスペクトルを前処理します:
[ ]:
# convert the spectra pandas.DataFrame to numpy.ndarray
spectra_test_np = spectra_test.to_numpy()
# preprocess the spectra
spectra_test_preprocessed = preprocessing.transform(spectra_test_np)
最後に、PLSモデルを使用してグルコース濃度を予測できます:
[ ]:
# predict the glucose concentrations
glucose_test_pred = pls.predict(spectra_test_preprocessed)
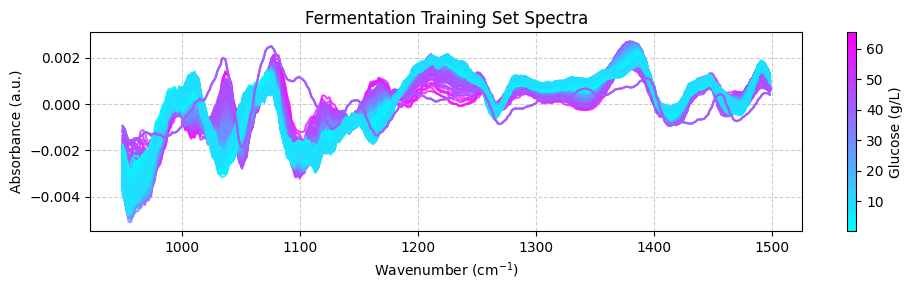
予測値を使用して、予測されたグルコース濃度に応じて色分けされたスペクトルをプロットできます:
[ ]:
plot_spectra(spectra_test_preprocessed, wavenumbers_cut, glucose_test_pred)
予測されたグルコース濃度をオフラインHPLC測定値と比較できます。時間経過に伴う予測とオフライン測定値をプロットします。各スペクトルは1.25分ごとに取得され、オフライン測定値は60分ごとに取得されました。
[ ]:
# make linspace of length of predictoins
time = (
np.linspace(
0,
len(glucose_test_pred),
len(glucose_test_pred),
)
* 1.25
/ 60
)
# plot the predictions
fig, ax = plt.subplots(figsize=(10, 3))
ax.plot(time, glucose_test_pred, color="blue", label="Predicted")
ax.plot(
hplc_test.index, hplc_test["glucose"] + 4, "o", color="fuchsia", label="Measured"
)
ax.set_xlabel("Time (h)")
ax.set_ylabel("Glucose (g/L)")
ax.set_title("Fermentation test set")
ax.legend()
ax.grid(True, linestyle="--", alpha=0.6)
fig.tight_layout()
plt.show()
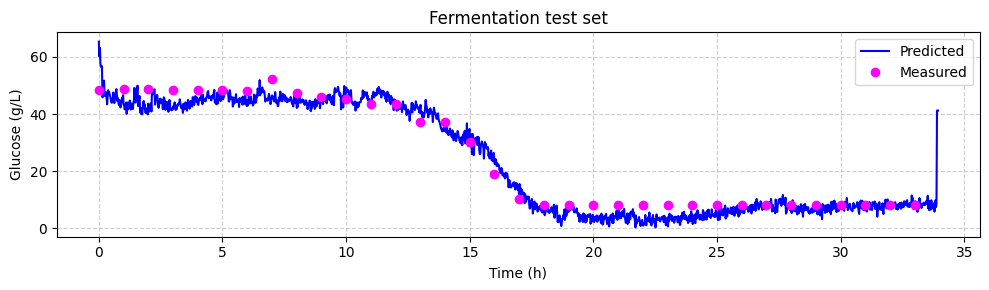
まとめ#
このチュートリアルでは、発酵データセットを使用して分光データ分析に機械学習を適用する方法を探求しました。目標は、リグノセルロース系エタノール発酵中のグルコース濃度を予測できる回帰モデルを構築することでした。カバーした内容の簡単な要約は次のとおりです:
はじめに:減衰全反射中赤外分光法(ATR-MIR)を介して収集されたスペクトルデータと対応するHPLC参照データを含む発酵データセットを紹介することから始めました。このデータセットは、リアルタイム発酵プロセスの研究に特に価値があります。
データのロードと探索:訓練データをロードし、その構造を調べ、スペクトル測定とHPLC測定の両方を詳しく見ました。この段階でデータを理解することは、成功した分析の重要な基盤です。
データの視覚化:視覚化を通じて、データセットのより明確な全体像を得ました。グルコース濃度で色分けされたスペクトルをプロットすることで、サンプル全体で化学的変動がどのように現れるかを観察できました。
データの前処理:範囲カット、線形補正、Savitzky-Golay導関数、標準スケーリングなどのいくつかの前処理技術を適用して、モデリング用のスペクトルデータを準備しました。これらのステップは、ノイズを軽減し、全体的なデータ品質を向上させるのに役立ちました。
モデルの訓練:処理されたデータを使用して、グルコース濃度を予測する部分最小二乗(PLS)回帰モデルを訓練しました。交差検証は、最適なコンポーネント数を決定し、モデルのパフォーマンスを評価するのに役立ちました。
テストと適用:最後に、訓練されたモデルをテストデータセットに適用して、リアルタイムでグルコース濃度を予測しました。これは、発酵中のグルコースレベルを追跡するためにモデルをどのように使用できるかを示しました。
このチュートリアルは、分光データ分析に機械学習を使用するための実用的な入門を提供します。これらのステップに従うことで、複雑なスペクトルデータから意味のある洞察を抽出し、実世界のアプリケーションを持つ予測モデルを構築できます。